Research Overview

Archaic Introgression and Human Population HistoryInferring the very ancient admixture events between Archaic hominins and us
|

Complex Traits Genetics in Recently Admixed PopulationsModeling changes in the genetic underpinnings of complex traits as a result of recent admixture
|
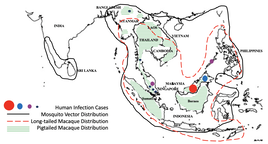
Dynamic Admixed Genomes in Non-human Study SystemsStudying the ongoing dynamics of admixture through the lens of fast evolving non-human species
|
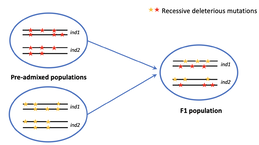
Methods Development in Population GeneticsMachine Learning and statistical methods development for inferring genomic parameters and signals
|